Selecting strandedness for RNA-seq
Last updated: June 3, 2025
Protocols can produce unstranded or stranded (forward/reverse) data
Overview
First-generation RNA-seq protocols did not retain the strand specificity of origin for each transcript, referred to as an unstranded protocol. However, it is now also possible to retain the strand information with a modified the RNA-seq protocol, known as strand-specific or stranded RNA-seq. For stranded protocols, directionality can be either forward or reverse.
Selecting strandedness in your RNA-seq pipeline
After you've uploaded the assay data and sample data to your RNA-seq experiment in Pluto, you'll be ready for the Configure pipeline step.
Along with the genome and library type, you'll need to select the strandedness from four options:
Auto
Forward
Reverse
Unstranded
.png)
Once you've selected the strandedness option for your data, click Run pipeline!
Once the pipeline runs (typically takes a few hours), you'll be able to see a visual confirmation of the strandedness in the report on the QC tab under the RSEM > Infer Experiment section.
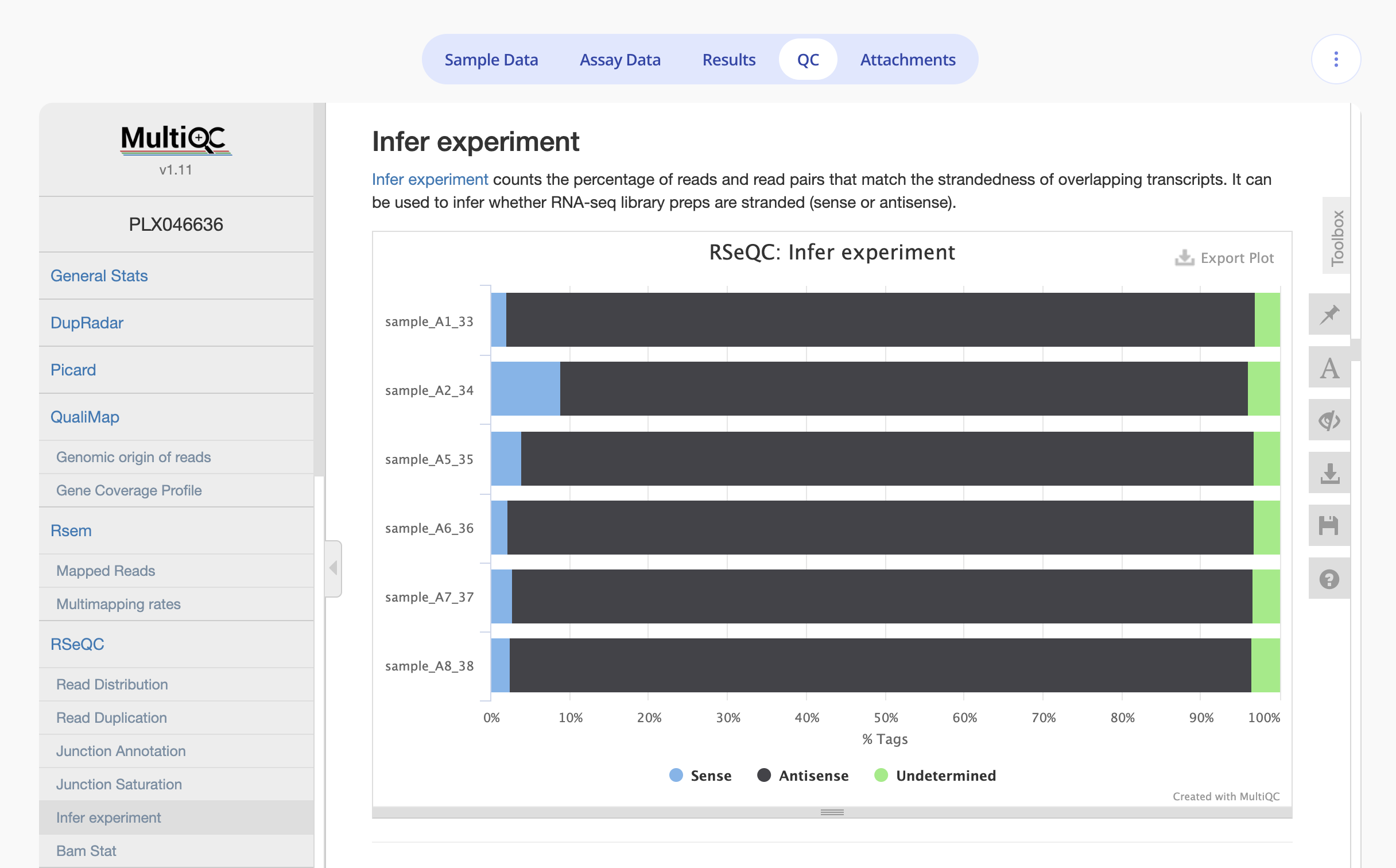
Example 1: reverse-stranded experiment
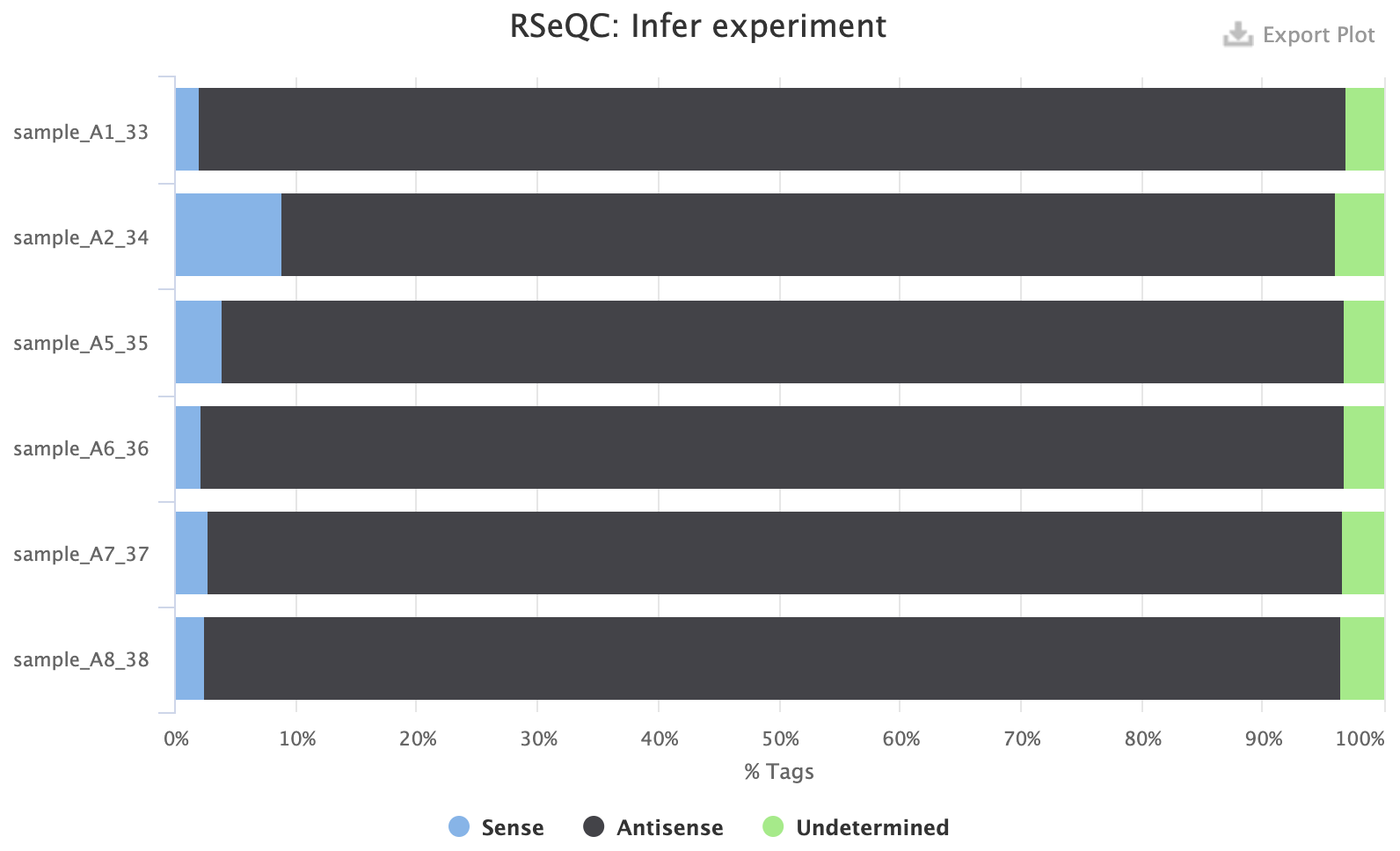
Example 2: forward-stranded experiment
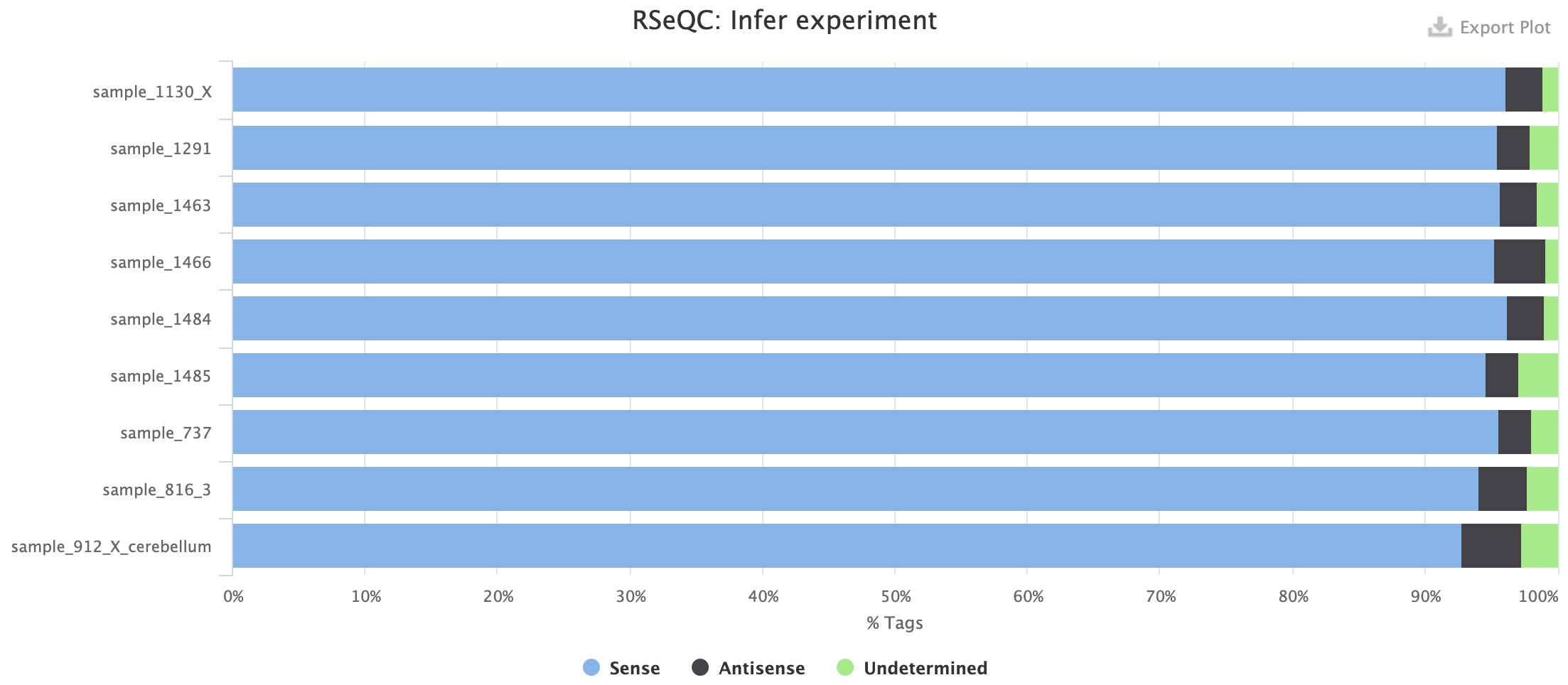
Example 3: unstranded experiment
What if I don't know the strandedness of my experiment?
No worries! Simply select Auto as the Strandedness option during the Configure pipeline step. The pipeline will automatically infer the correct strandedness for each sample in your experiment.
What if I selected the wrong strandedness of my experiment?
You can check if you selected the correct strandedness (or if you accidentally selected the wrong strandedness) of your experiment after the pipeline completes. When the pipeline completes, you'll receive an email notification letting you know that the QC is ready to review.
Once complete, go to the experiment and view the QC tab. If the strandedness initially selected was incorrect, you'll see a warning at the top indicating the strandedness that was used, and the strandedness that the pipeline inferred to be correct.

If you see the "Fail strandedness check" warning on your experiment, you can copy the experiment to rerun the pipeline. Check out the tutorial here.
(Once a copy has been made, you can archive the original experiment).
Run a new pipeline the same way as before, but using the correct inferred strandedness (or the Auto option). When the pipeline completes, you'll receive an email notification. Confirm that the new experiment QC looks good (has the correct strandedness) and you'll be ready to do your analysis!