Introduction to RNA-seq
Last updated: June 5, 2025
RNA sequencing (RNA-seq) is a popular method for measuring gene expression
Overview
When creating an experiment on Pluto, there is an option to create a “Bulk RNA-seq” type of experiment. This article will teach you about short-read bulk RNA-seq and why it’s important.
Why measure RNA?
Expression of a particular RNA generally indicates that the cell/organism needs the instructions that that particular RNA contains. Whether it is a messenger RNA (mRNA) which encodes a particular protein the cell currently needs or a non-coding RNA which can have many functions within the cell that expresses it, RNAs are critical for proper cell function. For example, if a cell receives a drug treatment it will respond by up regulating or down regulating particular genes. These changes can be detected by measuring the level of RNA expression for those genes which can help us infer the specific effects that drug treatment is having on those cells. Therefore, measuring RNA expression levels can provide us with insightful details about the current molecular state of a cell or tissue of interest and allow us to better understand specific cellular processes.
How RNA-seq achieves this
Unlike previously developed tools which only allowed the detection of a subset of RNAs, RNA sequencing (RNA-seq) is an incredibly powerful tool which allows us to detect changes in ALL of the RNAs within the cells of interest.
This is extremely important because it eliminates any bias and instead provides us with all the information available regardless of our prior knowledge. This is an extremely important technological advance which allows us to learn details about the development of an organism or the development and progression of disease that were previously unattainable.
Below is a timeline highlighting the various technological advances that lead to the tools we use today
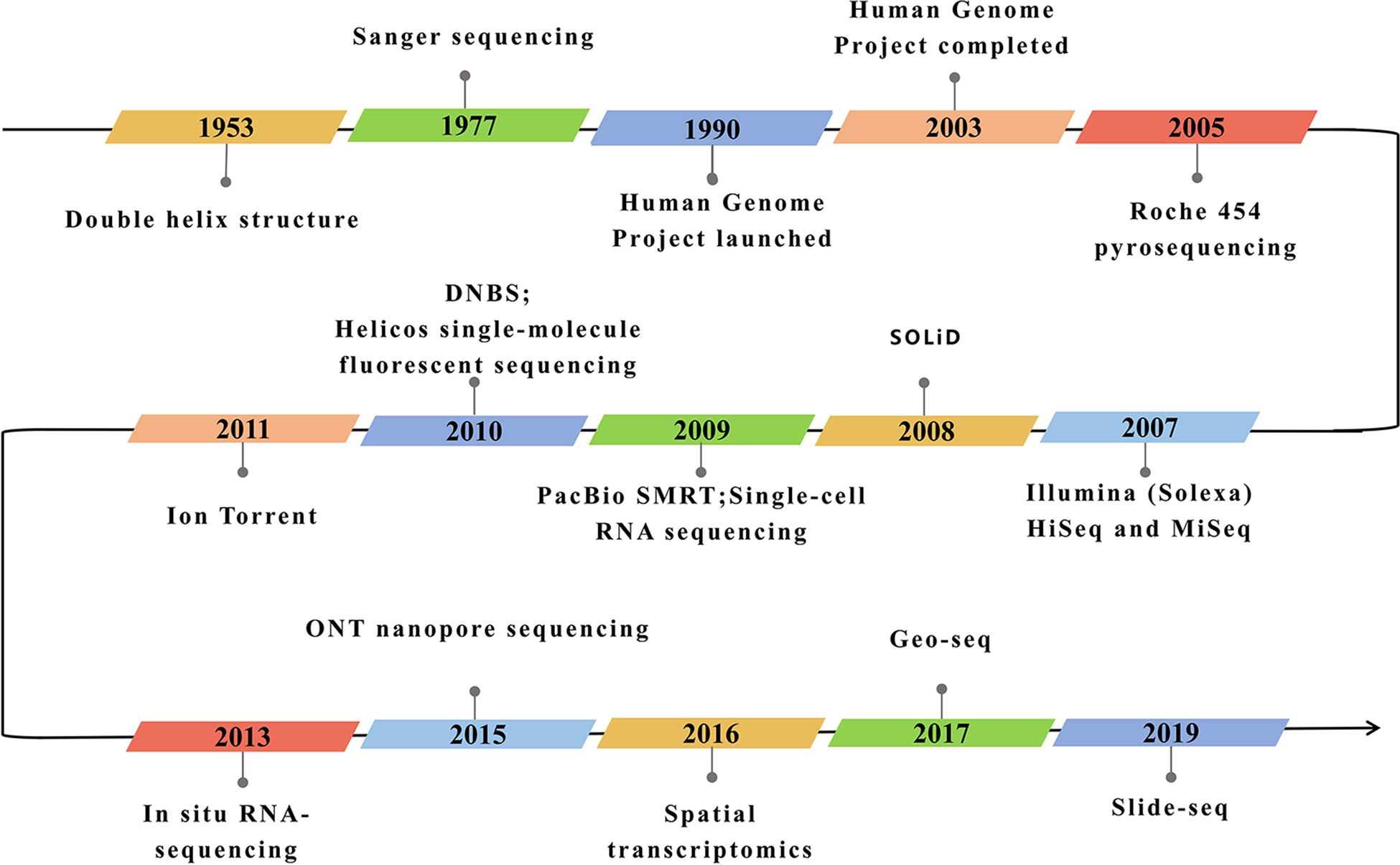
Image from Hong et al 2020
Things to consider when designing your bulk RNA-seq experiment
Should you use Oligo-dTs which will select for processed mRNAs by binding to their Poly-A tail or randomoligonucleotides which will bind to all RNAs irrespective of their type and processing status
Removal of ribosomal RNAs(rRNAs) - there are various kits used to achieve this. It is generally important to do since rRNAs are abundant and can therefore contaminate your sample
Perform single or pair-ended sequencing - The single stranded method is cheaper and faster because it only sequences the cDNA from one end whilst the pair-end method sequences the cDNA from both ends thus providing more in depth coverage
Stranded vs not stranded library preparation - although mRNAs are single stranded and have polarity (5’ and 3’ ends), this information is lost after cDNA synthesis during a typical library preparation, however there are a number of methods for creating a stranded library which retains this information and can be useful in interpreting the final results and are thus recommended
Depth of coverage - for a basic view into expression differences of highly expressed genes from an organism which has a reference genome 5 million reads provides enough information. If one seeks to find more detailed information about expression such as isoform differences, lowly expressed genes, small RNA details 20-50million reads are more appropriate.
Replicates - it is currently accepted to perform a minimum of 3 biological replicates per condition.
Importantly, when designing an RNAseq experiment it is important to know exactly what question you want answered in order to ensure best quality results. If you need help deciding on any of these variables we are happy to assist you. - CONTACT US link? - or a CHAT link?
Below is a comparison table of all the technologies currently available.
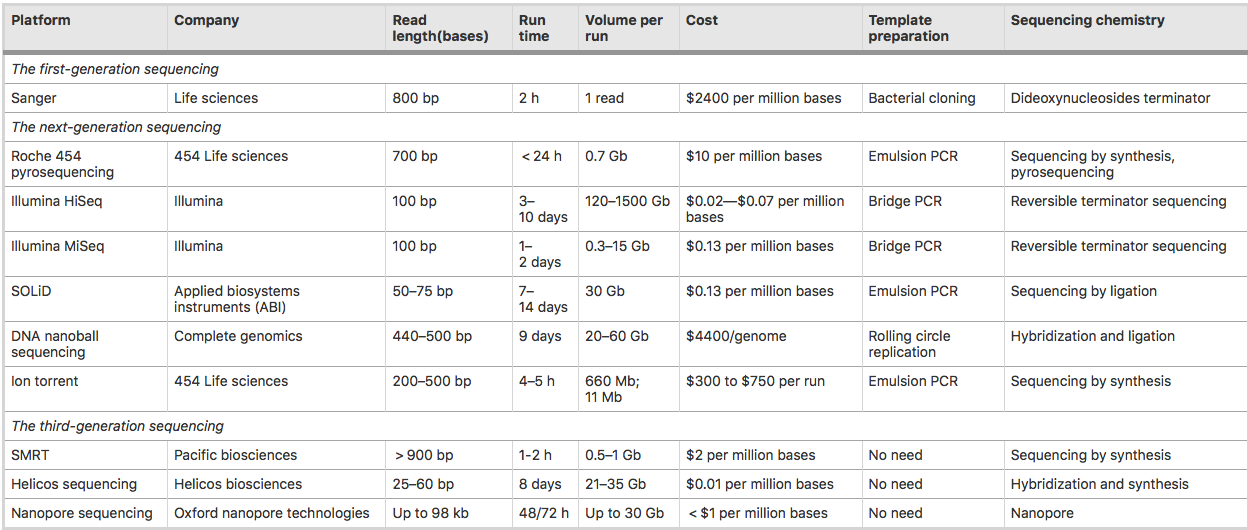
Image from Hong et al 2020
The next step is receiving the results from the sequencer and analyzing the data - raw sequencing files are called FASTQ and they need to undergo many bioinformatic steps of data quality checks and clean-up as well as final alignment to a reference genome. One can use a pseudo-aligner such as Kallisto which is fast or a splice-aware alignment tool which is significantly slower but delivers more detailed results. Pluto offers both versions and can deliver results in a few hours using the fast aligner and next day using the splice aware aligner. Details on the specific pipelines used can be found on our RNAseq analysis page (link here to another article).
Notable applications of RNA-seq
RNA-seq is an extremely powerful tool and can be used for a variety of applications. Some examples below but RNA-seq can help answer most questions one might have about gene expression changes. A short list of examples are provided below:
Biomarker identification for various diseases
Therapeutic response and disease variants (SNP identification)
Identification of developmental pathways
in vitro cell differentiation protocol development
Alternative splicing and isoform expression information
Biomarker identification for various diseases
Therapeutic response and disease variants (SNP identification)
Identification of developmental pathways
in vitro cell differentiation protocol development
Alternative splicing and isoform expression information
Due to the increased amount of RNA-seq datasets it is now possible to mine and compare previously published studies of interest therefore giving one the ability to place your own data into a larger context and provide a new layer of understanding and confidence in the data. Pluto has developed a large database that is easily searchable and where one can compare existing datasets in a fast and intuitive manner. Click here to start exploring. If your dataset of interest is not yet included in our database please send us an email with the GEO number and we will be happy to add it and let you know as soon as its available.
References and resources
Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat Rev Genet 20, 631–656 (2019). https://doi.org/10.1038/s41576-019-0150-2
Han, Y., Gao, S., Muegge, K., Zhang, W., & Zhou, B. (2015). Advanced Applications of RNA Sequencing and Challenges. Bioinformatics and biology insights, 9(Suppl 1), 29–46. https://doi.org/10.4137/BBI.S28991
Hong, M., Tao, S., Zhang, L. et al. RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol 13, 166 (2020). https://jhoonline.biomedcentral.com/articles/10.1186/s13045-020-01005-x